Making PyMOL Pretty - A Guide to Generating Illustrations
The goal of this exercise is to give some introductory experience in
generating high quality images that can be used in thesis
introductions, papers and so on.
Starting
You will need to download a PDB file from www.pdb.org
for editing in PyMOL. When you are at the site, type "hemoglobin" into
the search window. A number of options will come up as you scroll down
the window. For this exercise, let's use the file for
oxyhemoglobin that is available under the record 1HHO.
Click on 1HHO and you will be brought to the full record. You can look
this over, but all we really need are the model coordinates in
the PDB file.
You can see these in a browser window by clicking on the Display menu in the upper right
corner of the record window and then clicking on PDB File. A long text file will
show. It allows you to see the contents of the file. The most
important component is the list of atom positions for the protein
molecule listed in lines that start with the word ATOM.
Save the text file to your computer as "1HHO.pdb" and you'll have the
file necessary to work with for this exercise.
Opening PyMOL and Getting Acquainted
If your computer behaves like mine, you should be able to double click
on the 1HHO.pdb file you just saved and it will open in PyMOL.
But, you can also start the program first and then open 1HHO.pdb using
the File menu. You should see
something that looks like this:
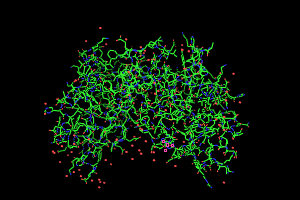
As an aside, this is only half of the
true hemoglobin molecule. That's often a problem - crystallography
often yields the smallest reproducible unit of a full protein molecule
and you need to generate the biological unit using crystallographic
symmetry. The symexp command allows you to do that. Here's how the command work in this instance.
I should also point out that there is a short cut. If you already know the name of your PDB file, you can use the fetch command in PyMOL. Check out the PyMOLwiki for how that works.
We will continue working without the symmetry mate. Before going
further, be sure to choose the type of mouse you are using from the Mouse menu. I
suggest using the Viewing Mode
for either 3-button or 1-button. The assignment of mouse actions is shown in the
lower right corner of the so-called Names Panel. Play with them a
little if you're unfamiliar with PyMOL.
To improve the initial image of the molecule, let's show the cartoon
image instead of the current line drawing. Again in the names panel, click on H (Hide)
button next to 1HHO and click on everything.
Then click on the S (Show)
button and click on cartoon.

A large green cartoon should appear on screen.
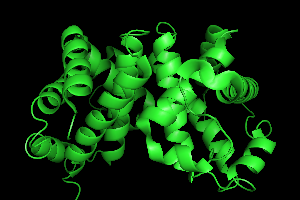
For the next section, it will help to see more of the model, so under Hide menu,
click everything
and under the Show
menu click Sticks.
Color
This is OK. But I like color. Color is one of the most entertaining
things to play with in PyMOL. The simplest way to adjust it is
using the C (Color) button in the names panel (next to Show and Hide).
Try out by
chain (e.g. c) under by chain or
even better, chainbows,
which colors each chain from blue to red from its N-terminus to its
C-terminus.
There are also a large number of blues, oranges, etc. that you can
color the entire molecule, but as we'll see that isn't often useful.
Instead, I tend to color selectively, and I do it all by command line
entry.
Also note, it can help in some cases to see the sequence of your
protein as you're working with it. I won't use this feature myself, but
if you're curious, click on Sequence in the Display menu.
If you are only interested in
working at the level of cartoon diagrams, this is a good place to break
off. This page
will give you some options in generating full cartoon diagrams of
proteins.
For a more advanced bit of cartooning, you can also align multiple structures. Check out this page for how.
To go further, you need to learn a bit about selecting specific parts
of the model in PyMOL. The levels of identification are:
- chain
- chain identifier (A, B, etc.)
- resn
- residue names. For protein (ALA, CYS, etc). For DNA (DA, DC, etc.)
for ligands it varies. In 1HHO, HEM is a ligand.
- resi
- residue number from the sequence
- name
- atom names (CA, CB, N, O, etc)
- elem
- elements (C, O, N, FE, etc)
So, for example - let's say I want to color atoms by element. There is
a simple way to do that under the C (Color) menu in the Names Panel, or
I can do it manually by entering a series of commands in by hand.

color red, elem o
color blue, elem n
color yellow, elem c
color orange, elem s
color magenta, elem fe
This colors each element in the molecule by a basic scheme. That's a
pretty boring set of colors, and PyMOL lets you go pretty wild. Using
the pallet of Colors
that
you can check out under the Settings menu,
there are too many options by count. But let's say I wanted to color
the carbon atoms of each chain distinctly, I'd do it this way:
color samarium, elem c and chain A
color antimony, elem c and chain B
This is my first use of selection algebra (covered intensely on the
PyMOLwiki). I am telling PyMOL that I want all atoms meeting the dual
criteria of being carbon AND on chain A to be colored "samarium".
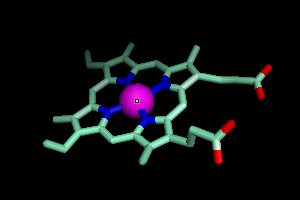
Your screen should now look a little like this.
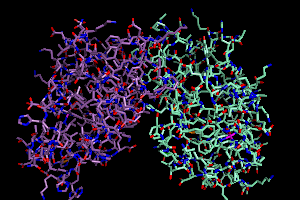
A Narrower Focus
This is obviously a pretty complicated picture, so let's say you just
wanted to see the part of hemoglobin where oxygen binds. There
are a lot of ways to get this, but here's one strategy.
First, let's create an "object" that includes only the heme from one
chain. The "select" command is huge for this stuff.
select heme,
resn HEM and chain a
The names panel will show your new object.

To focus on that, go to 1HHO and Hide Everything.
Then, next to (heme),
Show Sticks.
and under the A
(Action) button, click Zoom. You
should see something like this.
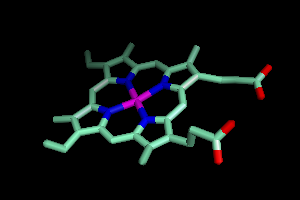
Better, but I like to show metal atoms as small balls, so I will create
a new selection, iron, that is only the iron atom from that heme and
make it a sphere.
select iron,
heme and elem fe
Note that I can now use "heme" as an identifier in my object and the
new object iron will only contain those atoms that are simultaneously
part of heme and are iron.
Next to the (iron) object, click on Show Spheres. And see what
you get. Too big for my tastes. But you can define the size of spheres.
alter iron, vdw=1.2
rebuild iron
Ah, better. The alter/vdw
command gives you a sphere of 1.2 Å.
Having gone this far, time to back up the work. Under the File menu, select "Save Session As...". This will save with a "pse"
extension. That file can be opened any time in the future. Note you
need to save separate back up files as you go. You can't do
simple update "Save" as in Word.
Adding Details
An isolated heme group is of little interest, but seeing it in the
context of the protein might add some information. Let's start by
looking at the environment around the iron atom, which is the
functional center of heme. To find all groups bonded to iron, we will
create a new selection that isolates groups that are within 3 Å of iron.
select febind, byres(iron expand 3) and
(not heme)
Let's break down the meaning of that line:
"iron expand 3" is all atoms within 3 Å of the iron atom
"byres(iron expand 3)" is all residues
within 3 Å of the iron atom
"and (not heme)" says, we already have the heme atoms saved elsewhere,
so let's leave them out.
OK, so now show what you've found. Next to febind, click Show Sticks. And to distinguish protein atoms from heme atoms, let's color the heme carbon atoms differently.
color pink, elem c and heme
Here's what you should see, with a little rotation using the mouse.
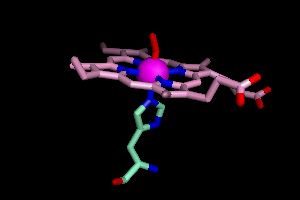
It turns out a histidine residue is attached to the iron, and so is a
molecule of dioxygen (oxyhemoglobin, remember?).
Let's say I wanted to show some amino acid side chains that surround
the O2 binding pocket. This isn't the most elegant way, but it
will work. Under 1HHO, Show Lines. You will see all the amino acids surrounding the O2 group.
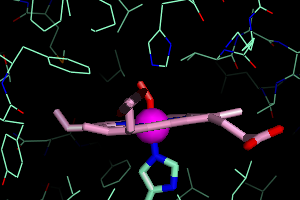
By right clicking on any residue that appears close to the O2, you can
get its residue number. For example, the Histidine residue above the O2
returns /1HHO//A/His`58/. That means it is His58 on the A chain. I also find that Leu29 and Val62 are in close proximity.
I'll create another selection that includes those three residues.
select o2bind, resi 29+58+62 and chain a
The algebra is a little more complex. I specified the three
residue numbers linked by "+" and then said that they must also be part
of chain a.
Go ahead an
show those by clicking Show Sticks
next to o2bind and Hide Lines next to 1HHO to clean things up.
Let's look further at His58. It is not bonded to the
dioxygen, but I'd like to put a dotted line between them to show the
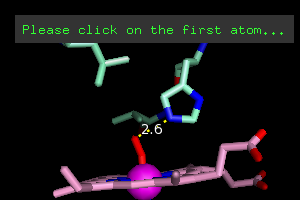
interaction. Under the Wizards menu, select Measurement.
It will tell you to click (left button) on the first atom. Click
on the blue nitrogen atom closest to the dioxygen. It will then
ask for the second atom, so click on the nearest oxygen. If you
got it right, you'll see this.
If you got that, you can click Done in the Names Panel. If not, click on Delete Last Object and try again, finally clicking Done when you've got it.
You've also got a new object in the Names Panel, (measure01). Click on Hide Labels, next to (measure01)
to get rid of the "2.6". Why? I don't like numbers in my
pictures, but you might... I also like a darker color for the dots, so
you can do that as you like - either type in the command line or use the C button in the Panel next to measure01.
color orange, measure01
Now, let's fill in the rest of the protein as a cartoon. Next to 1HHO, Show Cartoon.
So now we're almost ready to take it home (maybe a good time to save
your work). To put this image in a document, the black background
will make it hard to see for the reader, so change the background to
white:
bg_color white
Gee. The cartoons take up a lot of the image. I'm going to de-emphasize them by making them partly transparent.
set cartoon_transparency=0.5
And you know, those backbond atoms are ugly...
hide (name c+o+n)
Note that you can select multiples of anything using the "+" linker. For example, I could do several residues by number (resi 7+77+102+103) or by name (resn ALA+CYS+MET).
And now I'm feeling OK about this picture. I get the orientation I want... and let's save it in case I lose my place.
view 1, store
From here on out, I can type "view 1" and I can get back this orientation.
Finally, there is one last piece. The image you would get if you
save it now would give lots of jaggies, so I will do two things to
improve resolution: (a) increase the image size to 900x900 pixels (or
whatever you like), and (b) raytrace the image;
viewport 900, 900
ray
Or alternately, if you want to do it in one step (doesn't give you a chance to see the image in the new window):
ray 900, 900
You can't rotate this image, but it's ready to print! Simply select "Save Image As" under the File menu and pick PNG as the output format. Ta Da!
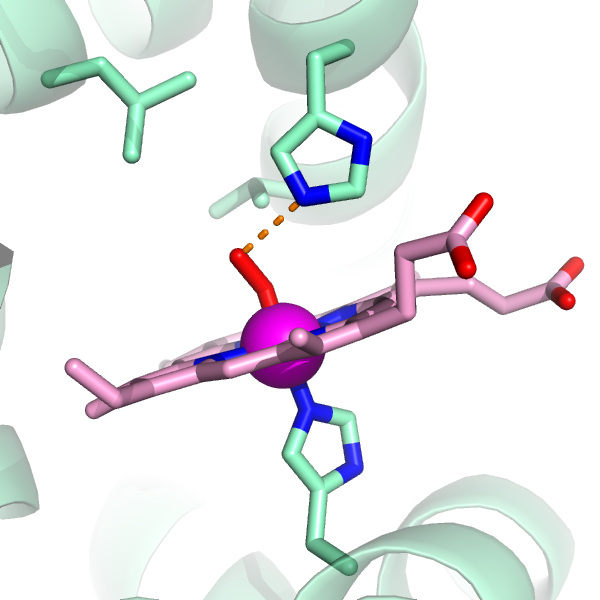
For my purposes, if I want to add labels, I import the image file
into Powerpoint and then create a slide that has residue names and so
on. It can be done in PyMOL, but I have not liked the results myself
(check under the Label menus in the panel to see your options.)
From here on out it gets harder. You need to start getting
frustrated with the software instead of my instructions. But one way or
the other, the PyMOLwiki will show you the way. Good luck.